Scientists have developed a powerful new computational framework that significantly accelerates drug discovery by enabling accurate simulation of rare molecular events without distorting their natural physics. The method, called PathGennie, represents a major advance in computer aided drug discovery by allowing researchers to predict how drug molecules disengage from their protein targets, a process that is central to understanding drug efficacy and safety.
The breakthrough has been reported in the Journal of Chemical Theory and Computation and has been developed by researchers at the S N Bose National Centre for Basic Sciences in Kolkata, an autonomous institute under the Department of Science and Technology. The software has been released as an open source tool, making it freely accessible to researchers worldwide.
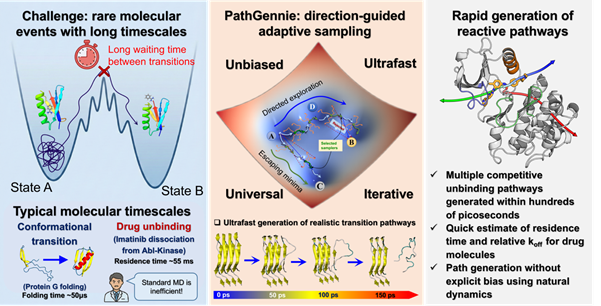
In modern pharmaceutical development, the effectiveness of a drug is not determined solely by how tightly it binds to a target protein, but also by how long it remains bound. This duration, known as residence time, is often a stronger predictor of therapeutic performance and side effects. However, accurately simulating the unbinding process is extremely challenging because it involves rare molecular events that occur on timescales of milliseconds to seconds. Conventional molecular dynamics simulations typically operate on much shorter timescales, making these events computationally prohibitive even on advanced supercomputers.
To overcome this limitation, existing approaches often rely on artificial biasing forces or elevated temperatures to accelerate the unbinding process. While effective in speeding up simulations, these techniques can distort the true physical pathways, leading to uncertainty in predicting how drugs behave under real biological conditions.
PathGennie introduces a fundamentally different approach. Instead of forcing molecular events to occur, it uses a direction guided adaptive sampling strategy that mimics natural selection at the molecular level. The algorithm initiates large numbers of extremely short, unbiased molecular dynamics trajectories, each lasting only a few femtoseconds. It then selectively extends only those trajectories that show meaningful progress toward a predefined end state, such as a drug exiting a protein binding pocket, while discarding unproductive ones.
This survival of the fittest strategy for molecular trajectories allows the framework to bypass the long waiting times associated with rare events without applying any artificial forces or temperature changes. As a result, the true kinetic pathways of molecular transitions are preserved, providing more reliable and physically accurate insights into drug behaviour.
The method is highly flexible and can operate in any set of collective variables, including high dimensional and machine learned coordinate spaces. By dynamically balancing exploration of new configurations with focused progression toward target states, PathGennie rapidly identifies transition pathways that would otherwise remain inaccessible through standard simulations.
In proof of concept studies, the research team demonstrated the capability of PathGennie to uncover multiple competing molecular pathways in complex systems. The framework successfully mapped how a benzene molecule escapes from the deep binding pocket of the T4 lysozyme enzyme, revealing several distinct exit routes. In another key example, the algorithm identified three separate dissociation pathways for the anti cancer drug imatinib as it unbinds from the Abl kinase protein. Notably, these pathways matched those previously reported using biased simulation techniques and experimental studies, validating the accuracy and reliability of the new method.
Beyond drug discovery, PathGennie is designed as a general purpose framework applicable to a wide range of rare event problems in chemistry and materials science. Potential applications include chemical reactions, catalytic mechanisms, phase transitions and self assembly processes. The framework is also compatible with modern machine learning approaches, allowing researchers to guide simulations using advanced data driven descriptors.
By making the software openly available, the developers have lowered the barrier for widespread adoption of this advanced technique. PathGennie is expected to play a significant role in accelerating drug discovery pipelines, reducing computational costs, and improving the reliability of predictions in molecular science, marking an important step forward in the use of computation to drive innovation in healthcare and beyond.

